
In a recent study posted to the bioRxiv* preprint server, researchers in the United States explored a novel technique to improve representation in viral genomic surveillance.
Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) evaded detection in the early phases of transmission due to the lack of regular viral genomic surveillance, allowing the virus to proliferate unchecked. In addition, poor surveillance over the ensuing months allowed SARS-CoV-2 variants to appear undetected. In light of this, minority groups have experienced a higher burden of coronavirus disease 2019 (COVID-19) cases and fatalities due to social and racial inequities.
 Study: A collaborative approach to improve representation in viral genomic surveillance. Image Credit: Studio.c / Shutterstock
Study: A collaborative approach to improve representation in viral genomic surveillance. Image Credit: Studio.c / Shutterstock
About the study
In the present study, researchers aimed to increase the capacity of microbial genome sequencing at universities using a new variant surveillance model.
From 22 July 2021 to 12 April 2022, six partner clinics associated with the study in Louisiana gathered 405 rapid antigen-positive clinical specimens from people living in nine parishes, including Allen, Bienville, Franklin, Jackson, Lincoln, Morehouse, Ouachita, Union, and Webster. More than 90% of the samples contained data related to demographics and vaccination status of the individuals.
In Georgia, Mercer University School of Medicine (MUSM) samples were gathered from Mercer Medicine (MM) community clinics and student health centers in the counties of Bibb, Chatham, DeKalb, and Fulton. If Polymerase chain reaction (PCR) analysis revealed a positive result, samples were sent for Illumina sequencing. The Jackson State University (JSU) Health Services Center in Mississippi served as the collection location. The Center supported the neighboring community's testing and immunization activities and acted as the principal COVID-19 testing facility for university students, staff, and visitors.
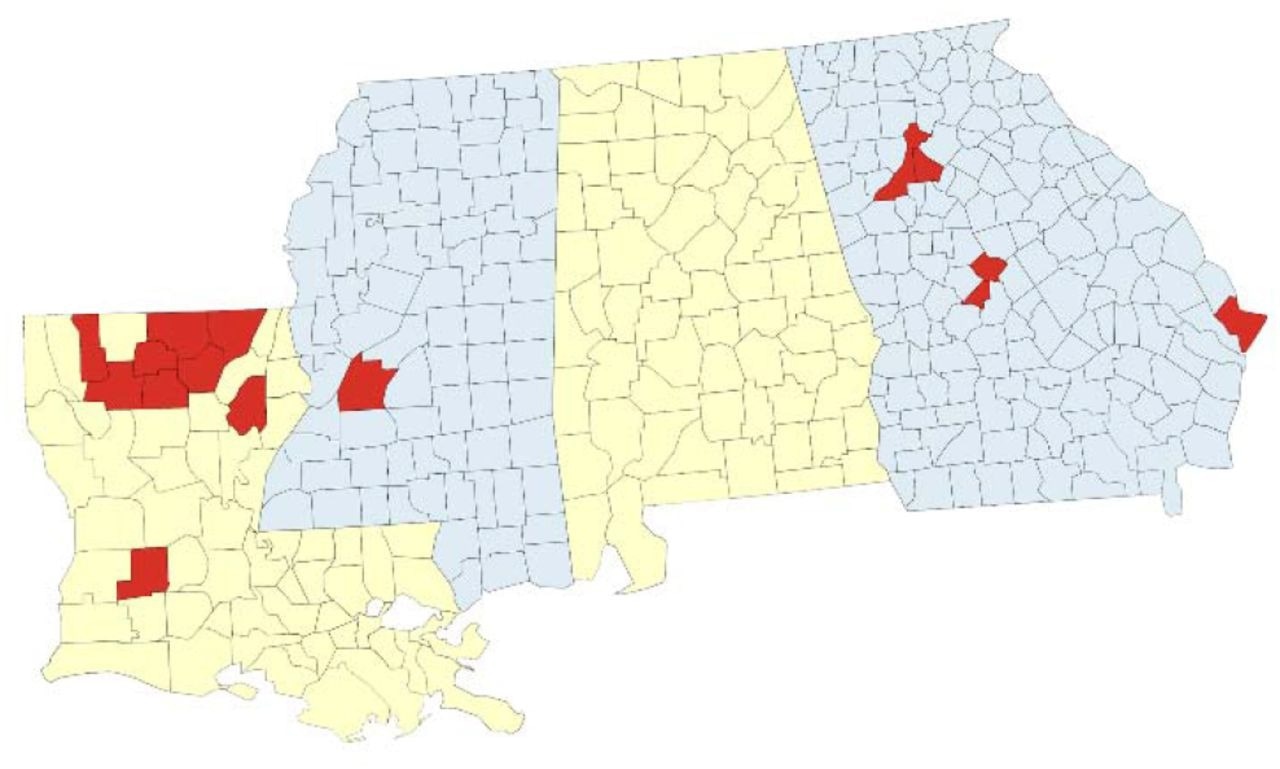 Geographic coverage of SARS-CoV-2 genomic surveillance in Louisiana, Georgia, and Mississippi. Map of the surveillance region with parishes (Louisiana) or counties (Georgia, Mississippi) where at least one specimen was sequenced by the network indicated in red.
Geographic coverage of SARS-CoV-2 genomic surveillance in Louisiana, Georgia, and Mississippi. Map of the surveillance region with parishes (Louisiana) or counties (Georgia, Mississippi) where at least one specimen was sequenced by the network indicated in red.
The team subsequently completed the surveillance loop by disseminating the variant data, weekly wastewater surveillance findings, and other COVID-19-related health data via a dashboard created for the people of north Louisiana. A matched subset of 16 SARS-CoV-2 positive samples was sequenced to verify the effectiveness of Nanopore MinION sequencing.
Results
Genetics & Genomics eBook

Non-White or people belonging to two or more races accounted for 60% of the study sample donors as opposed to 41% of the sample parishes. Despite representing just 3.1% of the population in northeast Louisiana, Hispanic donors accounted for 8.4% of specimens and 13.8% of the total cases reported by the Louisiana Department of Health. Hispanic individuals were also significantly more likely to develop COVID-19. The genomes were eventually retrieved from 272 PCR-positive specimens out of the 405 specimens collected in Louisiana, 389 of which showed detectable SARS-CoV-2 ribonucleic acid (RNA).
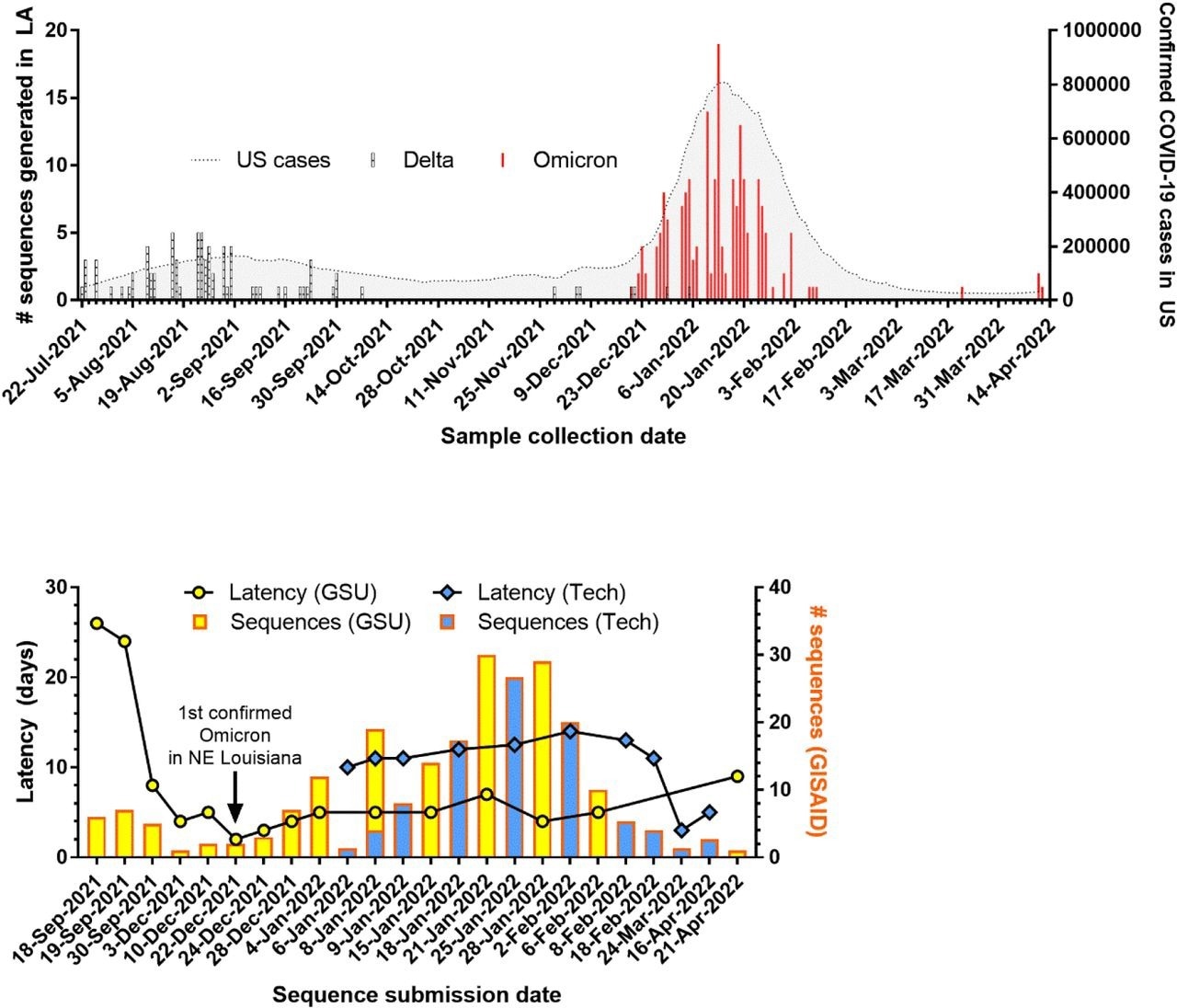 SARS-CoV-2 genomic surveillance in Louisiana. (A) Number of variants sequenced over time in Louisiana overlaid with number of confirmed COVID-19 cases in the U.S. (B) Number of sequences uploaded and latency for new sequencing entities established at Grambling State University (GSU) and Louisiana Tech University (Tech).
SARS-CoV-2 genomic surveillance in Louisiana. (A) Number of variants sequenced over time in Louisiana overlaid with number of confirmed COVID-19 cases in the U.S. (B) Number of sequences uploaded and latency for new sequencing entities established at Grambling State University (GSU) and Louisiana Tech University (Tech).
The team discovered two new BA.5 subvariants with immune escape mutations. With 100% consensus sequence identity observed in 13 of the 16 samples along with 99.9% sequence identity in three of 16 samples, the results demonstrated efficient accuracy of the Nanopore sequencing procedure and analysis pipeline. Within the gene that encoded the replicase auxiliary protein NSP13, the three samples shared the same discordant basecall, namely, T by Illumina sequencing and G by Nanopore sequencing at position 17,259. The reference SARS-CoV-2 genome also possessed a G at this location. The evidence for the E1264D mutation found in the samples analyzed using the Illumina methodology was significant. In homopolymeric tracts, nanopore basecalling accuracy declined, but it was unclear why this discordance was present.
The team discovered that the Omicron specimens' mean Cq values were considerably more significant than those of the Delta specimens. This supported earlier observations that Omicron infections, as opposed to Delta infections, had lower viral gene copy numbers as indicated by real-time reverse transcription PCR (RT-PCR) or lower infectious viral loads as determined by focus-forming assay. However, this variation was not statistically significant. In addition, the mean Cq value of unvaccinated people with no prior illness was 1.06 cycles lower than that of completely vaccinated people who did or did not have a prior history of infection.
Overall, the study findings showed that fostering collaborations between academic teams and neighborhood health clinics and instituting genomic surveillance capacity at rural academic institutions resulted in an effective strategy for enhancing pandemic preparedness.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- A collaborative approach to improving representation in viral genomic surveillance, Paul Kim, Audrey Y. Kim, Jamie J. Newman, Eleonora Cella, Thomas C. Bishop, Peter J. Huwe, Olga N. Uchakina, Robert J. McKallip, Vance L. Mack, Marnie P. Hill, Ifedayo Victor Ogungbe, Olawale Adeyinka, Samuel Jones, Gregory Ware, Jennifer L. Carroll, Jarrod F. Sawyer, Kenneth H. Densmore, Michael Foster, Lescia Valmond, John Thomas, Taj Azarian, Krista Queen, Jeremy P. Kamil, bioRxiv 2022.10.19.512816, DOI: https://doi.org/10.1101/2022.10.19.512816, https://www.biorxiv.org/content/10.1101/2022.10.19.512816v2
Posted in: Medical Research News | Medical Condition News | Disease/Infection News
Tags: Antigen, Assay, Coronavirus, Coronavirus Disease COVID-19, covid-19, Gene, Genome, Genomic, Illumina, Immunization, Medicine, Mutation, Omicron, Pandemic, Polymerase, Polymerase Chain Reaction, Protein, Respiratory, Ribonucleic Acid, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, students, Syndrome, Transcription, Virus

Written by
Bhavana Kunkalikar
Bhavana Kunkalikar is a medical writer based in Goa, India. Her academic background is in Pharmaceutical sciences and she holds a Bachelor's degree in Pharmacy. Her educational background allowed her to foster an interest in anatomical and physiological sciences. Her college project work based on ‘The manifestations and causes of sickle cell anemia’ formed the stepping stone to a life-long fascination with human pathophysiology.
Source: Read Full Article