. DOI: 10.1126/scisignal.ade3599")
Memory B cells depend on autophagy for their survival, but the protein Rubicon is thought to hinder this process. Researchers from Osaka University have discovered a shorter isoform of Rubicon called RUBCN100, which enhances autophagy in B cells. Mice that lacked the longer isoform, RUBCN130, produced more memory B cells in a way that relied on autophagy. These findings provide further insight into the role of Rubicon in autophagy.
Autophagy is a mechanism that degrades and recycles damaged components or organelles in cells, which plays an important role in various systems. In the immune system, autophagy is involved in B cell self-renewal and the survival of memory and plasma B cells. However, the effects of enhancing autophagy in B cells are not well understood.
The protein Rubicon (RUBCN) inhibits autophagy and impacts various conditions like fatty liver, kidney fibrosis, brain α-synuclein accumulation, systemic fat atrophy, osteoporosis, and myocardial ischemia/reperfusion injury. However, RUBCN deficiency can also lead to metabolic syndrome and decreased Sertoli cell function in mice.
RUBCN also regulates noncanonical autophagic processes like LC-3–associated phagocytosis (LAP), LC-3–associated endocytosis (LANDO), or micropinocytosis (LAM). Loss of RUBCN impairs LAP/LANDO and enhances fungal infection and neurodegeneration, but enhances antitumor “T cell” responses and germinal center (GC) B (BGC)–cell differentiation in mouse models. However, the exact mechanisms of RUBCN’s control over autophagy and other functions are still not fully understood.
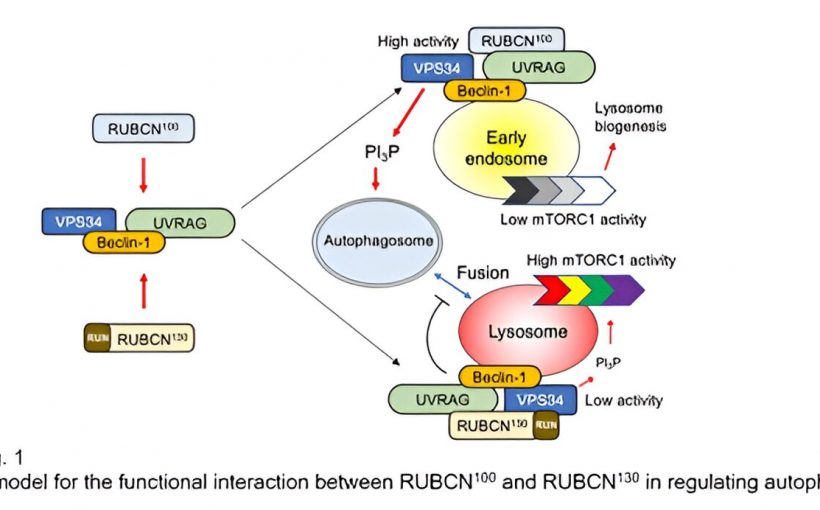
In a study published in Science Signalingresearchers have identified and characterized a RUBCN isoform called RUBCN100. RUBCN100 is translated from alternative start codons of the same messenger RNA that encodes RUBCN130. Autophagy is enhanced in B cells deficient in RUBCN130 or expressing RUBCN100.
In mice, selective deficiency of RUBCN130 enhances the generation of memory B cells and suppresses the differentiation of plasmablasts. The increase in B cell memory is further increased by enhanced recall responses in B cell-specific RUBCN knockout (RUBCN B-KO) mice. Importantly, elevated canonical autophagy alters the fate of BGC cell differentiation without affecting GC formation or cell survival in RUBCN B-KO mice.
The presence or absence of the RUN domain in RUBCN isoforms may explain their distinct functions in regulating autophagy. RUBCN100-VPS34 complexes enhance VPS34 activity, promoting autophagosome formation. On the other hand, RUBCN130-VPS34 complexes suppress autophagy by inhibiting VPS34 activity and autophagosome-lysosome fusion. Additionally, RUBCN100 restrains mTORC1 in early endosomes, delaying its activation. Put simply, RUBCN100 promotes autophagy, while RUBCN130 has a suppressive role.
In summary, the team found a RUBCN isoform called RUBCN100 and showed that the interaction between the two RUBCN isoforms is important for regulating autophagy. The balance of these isoforms is crucial for maintaining cellular balance and controlling autophagy and mTORC1 activity. A deficiency in RUBCN130 and enhancing autophagy with drugs can increase memory B cell generation. The discovery of RUBCN100 not only resolves conflicting findings about RUBCN’s functions but also sheds light on manipulating B cell memory.
More information:
Chao-Yuan Tsai et al, Opposing roles of RUBCN isoforms in autophagy and memory B cell generation, Science Signaling (2023). DOI: 10.1126/scisignal.ade3599
Journal information:
Science Signaling
Source: Read Full Article